Hoy, 23 de abril de 2026, la FDA aprobó Otarmeni™ (lunsotogene parvec-cwha), desarrollado por Regeneron Pharmaceuticals. Es la primera terapia génica para pérdida auditiva genética causada por variantes bialélicas en el gen OTOF, indicada para pacientes pediátricos y adultos con pérdida auditiva sensorineural severa a profunda (>90 dB HL).
¿Qué falla en estos pacientes?
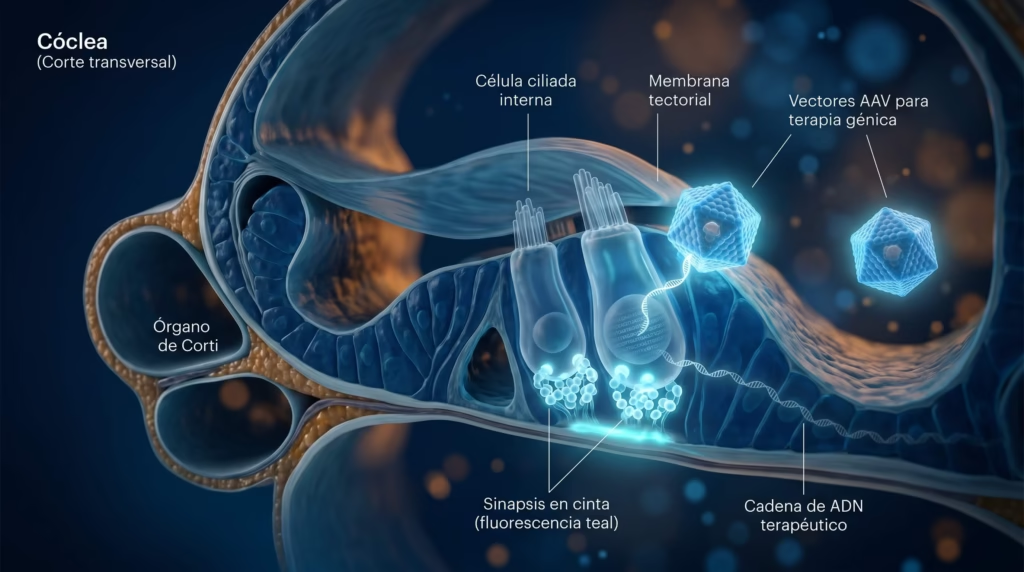
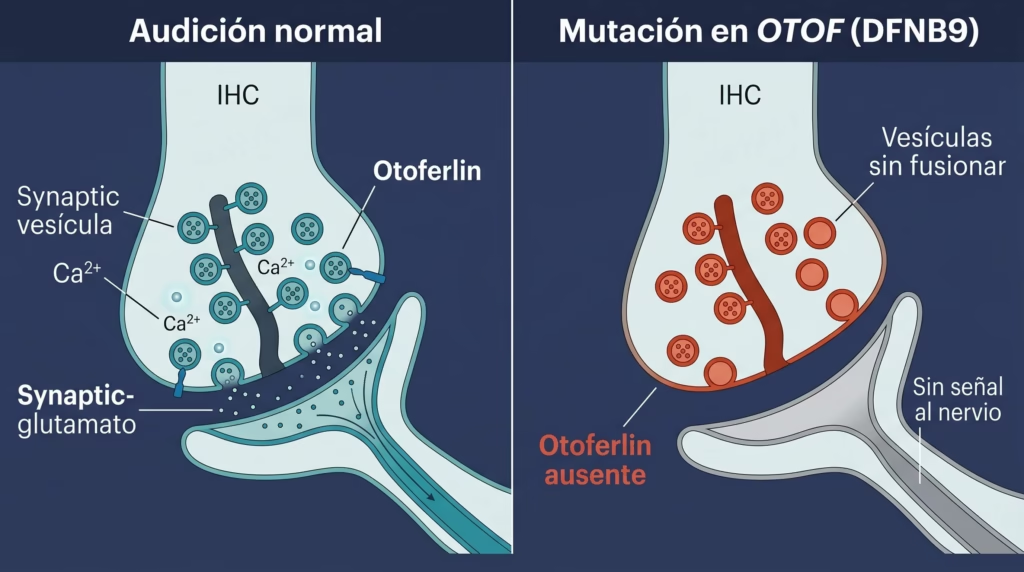
El gen OTOF codifica otoferlin, una proteína que actúa como sensor de calcio en las células ciliadas internas (IHCs) de la cóclea. Su función es disparar la fusión de vesículas sinápticas para liberar glutamato en las sinapsis en cinta (ribbon synapses): el paso que convierte la señal mecánica del sonido en impulso nervioso hacia el cerebro. Sin otoferlin, el oído detecta el sonido, pero la señal nunca llega al cerebro.
Las células ciliadas externas funcionan con normalidad, por lo que las pruebas de emisiones otoacústicas pueden salir normales en recién nacidos, lo que complica el diagnóstico temprano. Las variantes en OTOF son responsables del 1–8% de la sordera congénita no sindrómica y constituyen la principal causa de neuropatía auditiva de origen genético.
Sin otoferlin, el oído “escucha” — las células ciliadas responden al sonido — pero el mensaje nunca llega al nervio auditivo. Es como cortar el cable entre el micrófono y el amplificador.
¿Por qué fue técnicamente difícil hacer esta terapia?
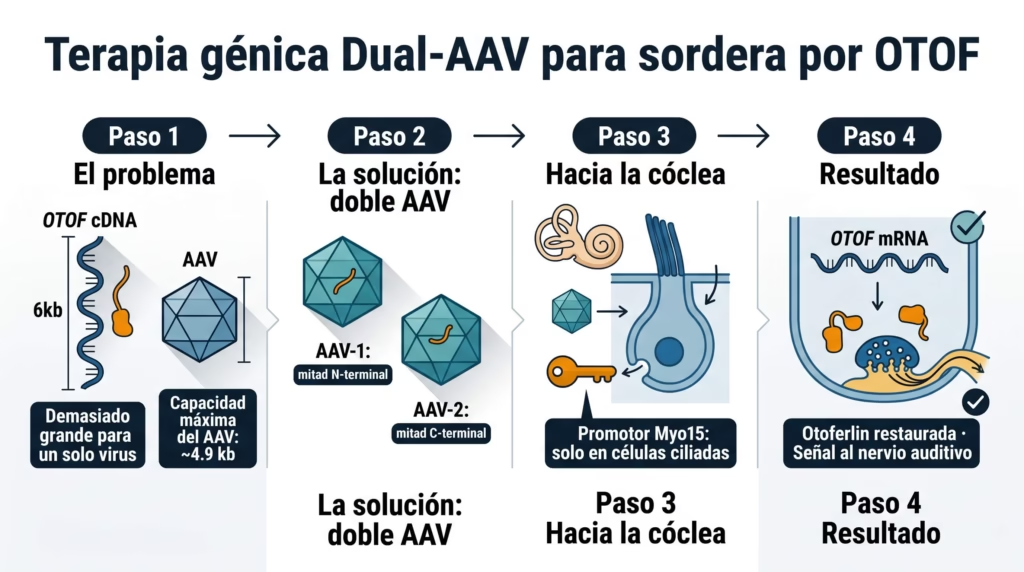
La otoferlin es una proteína grande. Su cDNA tiene ~6 kb, lo que excede la capacidad de carga de un solo vector viral (AAV), que es la herramienta estándar para terapia génica. Un solo AAV carga como máximo ~4.9 kb. La solución fue usar una estrategia de doble vector AAV (dual-AAV): dos virus que, juntos, entregan la secuencia completa y la reconstituyen dentro de la célula.
El gen introducido está bajo el control de un promotor específico de células ciliadas (Myo15), diseñado para restringir la expresión únicamente a las células que normalmente producen otoferlin. El tratamiento se administra mediante infusión intracocllear bajo anestesia general, en un procedimiento similar al de un implante coclear.
Resultados del ensayo CHORD
El estudio CHORD (Fase I/II, multicéntrico, abierto) evaluó Otarmeni en niños con variantes en OTOF y sordera profunda (>90 dB HL). El punto primario de eficacia fue alcanzar un umbral auditivo ≤70 dB HL a las 24 semanas: un nivel que generalmente permite evitar el implante coclear y posibilita la audición acústica natural.

¿Por qué es importante este avance?
La sordera congénita afecta aproximadamente a 1 de cada 500 recién nacidos. En los casos causados por mutaciones en OTOF, los niños nacen sin la capacidad de escuchar nada — y el período más crítico para el desarrollo del lenguaje oral es precisamente los primeros dos años de vida. Cada mes sin audición en esa ventana tiene consecuencias directas en la adquisición del habla, la integración social y el desarrollo cognitivo.
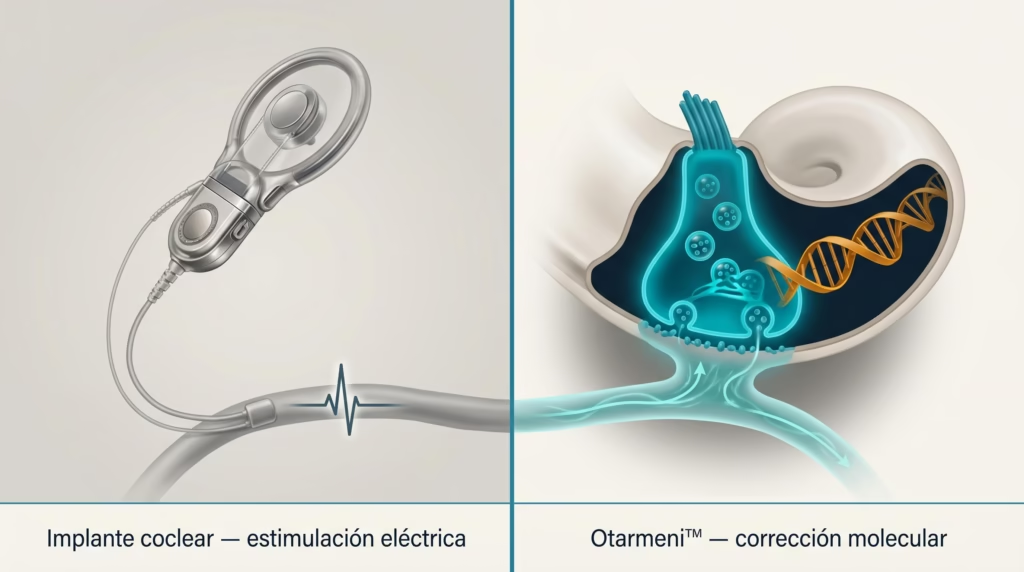
Hasta ahora, el estándar de atención era el implante coclear: un dispositivo que estimula directamente el nervio auditivo de forma eléctrica, sin pasar por las células ciliadas dañadas. Funciona, y los resultados en pacientes con DFNB9 son generalmente buenos. Pero tiene limitaciones reales: dificultad para distinguir tonos musicales, menor inteligibilidad en ambientes ruidosos, y la necesidad de un dispositivo externo permanente.
Lo que cambia con Otarmeni es fundamental: el niño escucha con su propio oído, procesando el sonido de forma fisiológica, no eléctrica. En el ensayo CHORD, varios participantes lograron audición dentro del rango normal — incluyendo escuchar un susurro. Eso no es algo que un implante coclear pueda garantizar.
Para una familia con un recién nacido diagnosticado con sordera por mutación en OTOF, esta aprobación significa que existe, por primera vez, una opción que puede restaurar la audición natural de forma duradera con una sola intervención — y en EE. UU., sin costo del medicamento.

Perspectiva
Según los investigadores del CHORD, esta aprobación representa una nueva etapa en el tratamiento de la sordera genética, donde restaurar la audición natural completa es ahora posible con una sola intervención. Antes de esto, el implante coclear era el estándar de atención: funciona, pero con limitaciones en el reconocimiento de tonos, música e inteligibilidad en entornos ruidosos.
Lo que hace diferente a Otarmeni es que no rodea el problema — corrige la causa molecular. Y hacerlo en un órgano tan pequeño y especializado como la cóclea, con un vector diseñado para no expresarse en ningún otro tipo celular, es un ejercicio de precisión que abre la puerta a terapias similares para otras formas de sordera genética.
Un punto que vale la pena resaltar: Regeneron ha decidido proporcionar Otarmeni de forma gratuita en los Estados Unidos a todos los pacientes clínicamente elegibles. En un campo donde los tratamientos de terapia génica frecuentemente alcanzan costos de millones de dólares, esta decisión es inusual. Los costos de administración del procedimiento (anestesia, quirófano) siguen siendo responsabilidad del paciente, pero el medicamento en sí no tendrá precio.
La aprobación acelerada llegó 61 días después de presentado el BLA, bajo el programa piloto National Priority Voucher (CNPV) de la FDA — y según los datos publicados, sin eventos adversos graves reportados en el ensayo clínico.
Referencias
- FDA Press Release, abril 23, 2026. FDA approves first-ever gene therapy for treatment of genetic hearing loss. fda.gov
- Regeneron Press Release, abril 23, 2026. Otarmeni™ (lunsotogene parvec-cwha) Approved by FDA as First and Only Gene Therapy for Genetic Hearing Loss. investor.regeneron.com
- Shearer et al. New England Journal of Medicine, octubre 2025. DB-OTO Gene Therapy for Inherited Deafness. DOI: 10.1056/NEJMoa2400521
- Adam et al. GeneReviews®, NCBI Bookshelf. OTOF-Related Hearing Loss. ncbi.nlm.nih.gov/books/NBK1251
- Vona et al. Genes, 2020. The Many Faces of DFNB9: Relating OTOF Variants to Hearing Impairment. DOI: 10.3390/genes11121411